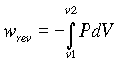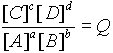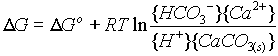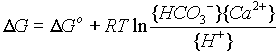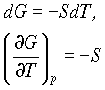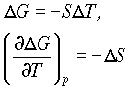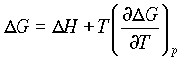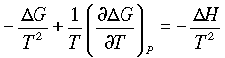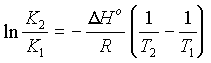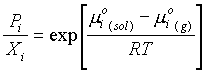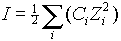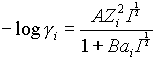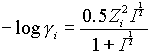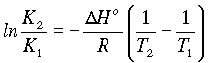dU=dq + dw
where U is the internal energy, q is the heat transferred to a system from the surroundings, and w is the work done on a system by the surroundings.
D
U=q + wdU = dq + dw
dU = dq - PdV
where PdV is the reversible expansion work done on the system by the surroundings.
sys and is the sum of changes inside the system, dSint, and entropy transferred to the system from its surroundings, dSsur.The second law is concerned with entropy (S), which is that part of the total energy of the system that is not available to do useful work. Entropy is a fundamental variable in thermodynamics, and is responsible for such phenomena as molecular diffusion. For an interesting view of entropy, check out the movies at The U.C. San Diego.
Statement One: "The total entropy change of the closed system is dS
dS
sys= dSint + dSsurdS
sur = dq/TFor a reversible process or equilibrium state of the system, the second law states that the internal entropy change is zero:
dS
int = 0For a spontaneous or natural process in the system
dS
int > 0So for any spontaneous natural process
dS
sys > dq/TThe internal entropy change for a closed system is zero at equilibrium and positive for a spontaneous process. The heat transferred to a closed system, divided by T, is equal to or less than the entropy increase for any possible process. The entropy of an isolated system increases in any spontaneous process and attains a maximum for any reversible process. (Stumm and Morgan, p. 19).
The Gibbs Free energy is that portion of the total system energy that is available for useful work.
dw=-PdV
At equilibrium, and with fixed composition (no addition of material):
dq = TdS(sys)
so
dU = TdS - PdV
So the internal energy is a function of entropy and volume. Another expression for total system energy is Enthalpy (H):
H = U + PV
dH = dU + PdV
When T and P are constant, we can use Gibbs expression for free energy
dG = dU + PdV - TdS
dG = dH - TodS
Where:
G = Gibbs free energy, kcal/mol
T = absolute temperature, oK
S = entropy, cal/oK
H = enthalpy, kcal/mol
This equation tells us if a reaction will happen, and in what direction.
a) Enthalpy: the total energy content of acompound (dH = dU+PdV) in kJ/mol; .
b) Free Energy: That part of the total energy that is available to perform "useful work", that is, other than PV work, such as electrical work, in kJ/mol.
Heat capacity is defined as the change in temperature of a system with a change of heat transferred to the system:
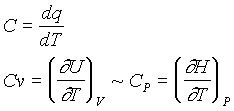
Heat capacity can be measured directly by calorimetry (at constant pressure), and can provide information on enthalpy for a closed-system process.
The driving force of a reaction is related to the concentrations of reactants and products.
aA + bB ---> cC + dD
At equilibrium this ratio is equal to the equilibrium constant, K.
The TOTAL FREE ENERGY is the sum of the free energies of each component.
G
T = nAGA + nBGB + nCGC + nDGDWhere Gi = Free energy / mole of i
ni = number of moles of i
DG = (SuiGi)products - (SuiGi)reactants
Where: ui = the stoichiometric coefficient
Gi= the free energy per mole
a) DG < 0, and GT decreases as the reaction proceeds, the reaction is spontaneous.
b) DG = 0, the reaction is at equilibrium, and GT is at a minimum
c) dG > 0, the reaction is not spontaneous as written, but proceeds in the opposite direction.
D
G = DG° + RT lnQWhere:
DG° = (SuiG°i)products - (SuiG°i)reactants
_
G°i = free energy per mole at standard state.
DG° = (SuiG°f,i)products - (SuiG°f,i)reactants
Gi =
DG°f,i + RT ln {i}Where {i} is the effective concentration or activity of species I
C
graphite + 3/2 O2 + Ca ---> Calcite DG°f = -269.78 kcal/mol
| Cgraphite + 3/2 O2 + Ca ----> Calcite | DG°f = -269.78 kcal/mol |
| H+ ------> H+ | D G°f = 0 |
| Ca-----> Ca++ | DG°f = -132.18 |
| H+ + C + 3/2 O2 ------> HCO3- | DG°f = -140.31 |
D
G° = (SviG°f,i)products - (SviG°f,i)reactants
CaCO3 + H+ --------> Ca++ + HCO3- G°r = -2.71
Reacting 1 mole of acid with one mole of calcite releases free energy, so the reaction is spontaneous.
The value of the activity of a substance is dependent on the choice of standard state conditions, or the conditions that result in unit activity.
6 For mixtures of liquids, (i) = Xi
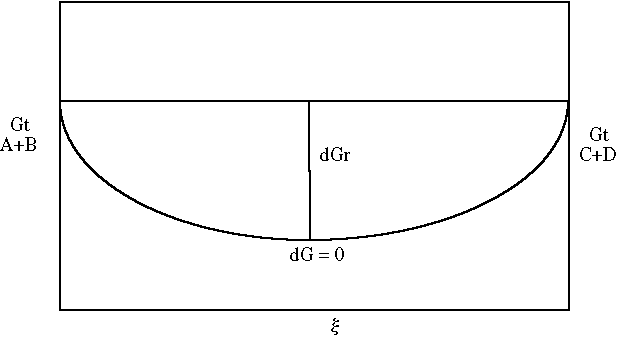
Returning to the example:
D
G = [DG°f,HCO3 + RTln{HCO3-} + DG°f,Ca++ + RTln{Ca++}] –Collecting Terms:
- [
DG°f,H+ + RTln{H+} + DG°f,CaCO3 + RTln(CaCO3)]W can state
and since the activity of a solid is unity:
D
G = DG° + RT ln QWhen the value of Q is identical to that of the equilibrium constant K, and
DG = 0,0=
DG° + RT ln K; DG°= -RT ln KWhen not at equilibrium, substituting we can get
D
G = -RT ln K + RTlnQ= +RT ln Q/K
when Q/K>1, not spontaneous
Q/K =1, equilibrium
Q/K < 1, spontaneous
Determine the equilibrium constant for the dissociation of water at 25°C:
H2O <----> H+ + OH-
1. Find the
DGf of the reactants and products:Species
DG°f,H2O -56.69
H+ 0
OH- -37.60
2. Sum the energies of formation to get a standard free energy of reaction
D
G° = (1)(0) + (1)(-37.6) - (1)(-56.69) = 19.09 kcal19.09 = -RTlnK
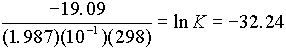
K = [H+]{OH-] = 10-
143. Use reaction rates to find equilibrium:
k1
H+ + OH- <----> H2O
k-1
with k1 = 1.4x1011 liter/mole sec
k2 = 2.5x10-5 /sec
·
These are the fastest rates known in aqueous solutions, where the reaction is essentially instantaneous, with the overall rate limited by the rate of diffusion of reactants.·
Since the reaction is elementary, we can write:Forward rate = k1[H+][OH-]
Reverse rate = k2[H2O]
·
Since at equilibrium, the forward rate = reverse rate:k1[H+][OH-] = k2[H2O]
k2[H2O]/k1 = [H+][OH-] (mole2/liter2)
·
Substituting for k's, and using 5.5 moles/l for water concentration: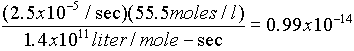
a) The chemical potential is the molar free energy G/ni, and describes the system for components not in the standard state, especially for components at concentrations other than 1 molar.
m
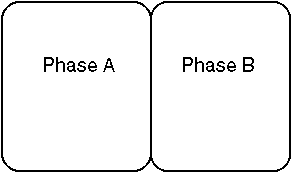
i = (dGi/dni)T,Pb) Example: Multi Phase system:
1) When two or more phases coexist at equilibrium, the chemical potential of all components in the system must be identical in each phase
at equilibrium,
mA = mB
·
if mA is greater than mB, than some reaction will occur to bring the system to equilibrium, ie mA = mB, resulting in the release of energy (-DG). Usually, this means the transfer of some component i from A to B.·
Any combination of phases will do.
The driving force of a reaction is
DG, as an expression of useful work (non-PV work). This is comprised of 2 components, enthalpy and entropy. At equilibrium:D
G = DH - T°DSSo the magnitude and direction of the driving force depends on the magnitude and direction of H and S. Neither by themselves controls the reaction.
a) Endothermic = positive DH
b) Exothermic = negative DH
D
H can be expressed in terms of G, S, and T as: DH° = DG° + T° DS° and represents the TOTAL energy of the system, both usable and unusable. At equilibrium, DH=+T°DS
DHo = S (DH°products) - S (DH°reactants)
dG = - SdT + VdP
Which at constant pressure can be written as
so the change in free energy with temperature is related to entropy. For a finite change in state:
divide through by T2:
and
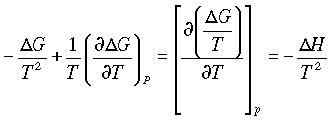
giving the Gibbs-Helmholtz equations
By differentiating the equation:
DG° = -RT lnK, we obtain
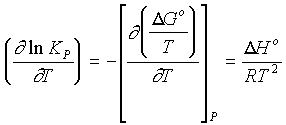
an equation called the Van't Hoff Isochore
So the equilibrium constant varies with temperature as a function of the Enthalpy of the system (Assuming dH does not vary with temperature.).
or
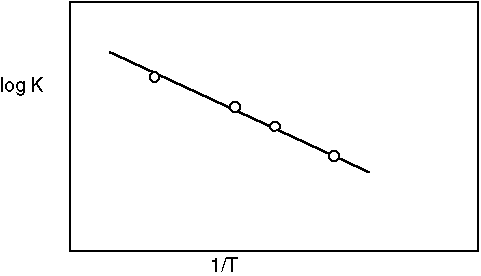
Plotting log K against 1/T results in a line whose slope is --DH°/2.3R
a) For an exothermic reaction, K must decrease as the temperature increases....
Chemical Potential: G = m = m(T,P)
m
= m°(T)
·
For n moles of pure gas at constant temperaturedG = VdP
·
From the ideal gas law:V = nRT1/P
so
dG = nRT dP/P
or
dG = nRTdlnP
·
Integrating from a standard pressure P° to a pressure P yieldsG-G°=nRTln P/P°
Dividing by n
G/n - G°/n = RTln P/P°
·
Since the chemical potential for a one-component phase is G/n:m
(g) - m°(g) = RTln P/P°When P = P°,
m(g) = m°(g), orm
i(g) = m°i(g) + RT lnP/P°If P° = 1
m
i(g) = m°i(g) + RT lnPand in an ideal gas mixture:
m
i(g) = m°i(g) + RT lnPi·
where the value of m at 1 atm is m° standard state, and Pi is the partial pressure of component i. If xi is the more fraction of component i: Pi = XiP, leading to:m
i(g) = m°i(g) + RT lnP + RTln Xia) This closely follows ideal gas mixtures: (159)
m
i(sol) = m°i(sol) + RT lnXiWhere: Xi = the mole fraction of i in solution
mi(sol) = mi(g)
mi(sol) + RT ln Xi = m°i(g) + RT lnPiRearranging
1) This equation states that the vapor pressure of component i is proportional to its mole fraction in a solution behaving ideally. This is the base equation for Raoults and Henrys law.:
The ideal gas vapor pressure of an ideal solution component is equal to the vapor pressure of the pure liquid component multiplied by the mole fraction of the component in the solution, or:
Pi = XiP°i
where Pi is the partial pressure,
Xi is the mole fraction, and
P°i is the partial pressure of pure component i.
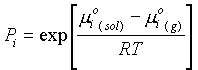
and when Xi = 1:
m°
i(sol) = m°i(g) + RT lnP°iThus the standard state chemical potential for the ideal solution component depends on the ideal gas state and the convention that Xi =1 is the standard state for the component in solution.
--For real solutions, Raoult's law behavior is approached as Xi ---> 1.
For a dilute solution the ideal gas vapor pressure of a volatile solute is proportional to its mole fraction in the solution; that is, escaping tendency of the solute molecule is proportional to their mole fraction, ie:
Pi = KiXi
This means that Ki is the slope of the plot of Pi vs Xi for sufficiently dilute solutions, or where solute-solute interactions are negligible.. So:
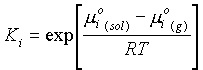 m°i(sol) for a Henrys law solute reflects the properties of a dilute solution, and for a Raoults law component it reflects the properties of a pure component, in other words, the solvent.
m°i(sol) for a Henrys law solute reflects the properties of a dilute solution, and for a Raoults law component it reflects the properties of a pure component, in other words, the solvent.For real solutions, Henry's law behavior is approached as Xi approaches 0.
Important:
1) The term activity is used for idealized concentration, vs actual concentration:
m
i = m°i + RT lnaiand
a
i = giXigiving the chemical potential in a real solution:
mi = m°i + RTlngiXiand
m
i = m°i + RTlngi+ RTlnXiWhere the expression RTln
gi is the partial molar free energy of the interactions that occur in nonideal mixtures.If mi(sol) = mi(g) then:
m
i(sol) + lngiXi = m°i(g) + RT ln PigfiIf the pressure of the vapor is low enough so that
gfi = 1, the solution-phase activity coefficient is:g1 = P1/P°1a
1 = g1X1 = P1/P°1
ai = gi Xi or {i} = g[i]
--The activity coefficient becomes less than one and the chemical equilibrium is affected, forcing the rewriting of the mass action equation to:
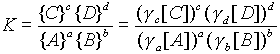
This describes the magnitude of the interionic effects, or the intensity of the electric field in a solution.
Compute the ionic strength of a solution containing the following concentration of ions:
[Ca2-] =10-4M, [CO32-] =10-5M, [HCO3-] =10-3M, [SO42-] =10-4M, [Na+] =10-3M
m =
([10-4 x 22]+[10-5x22]+10-3x12]+10-4x22]+[10-3x12])m =
1.43x10-3I ~ 2.5x10-5xTDS; I = 1.6x10-5 x specific Conductance
![]()
This is the simplest equation, and is good up only up to I = 0.005
·
Based on the effect ionic interactions should have on free energy. In an ionic solution, positive ions will tend to have a cloud of negative ions around them, and negative ions a cloud of positive ions.·
If it is assumed that the ions are point charges, the interactions are entirely electrostatic, and the ions around any particular ion follow a Boltzmann distribution.
Where:
A = a constant that relates to the solvent, and varies with temperature. (A = 1.82 x 106(eT)-3/2, e = dielectric constant.)
A = 0.5 at 15°C; 0.509 at 25°C
B = a constant that relates to the solvent and varies with temperature. B=50.3(eT)-1/2
B = 0.326x10
8 at 15°C; and 0.328x108 at 25°C.a
i =a constant that relates to the diameter of the hydrated ion. For monovalent ions except H+ this is about 3-4 x 10-8Zi = the charge of the ion
This approximation is useful in solutions of mixed ions with varying charges, since the limiting law is not satisfactory with mixed charge types, and is useful up to I = 0.1.
This is best at higher ionic strengths; to about I = 0.5m
![]()

·
All solutions are charge balanced, with the meq of anions exactly balanced by the meq of cations. The standard equation is: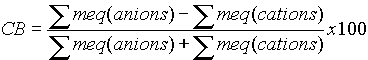
The usual QA cutoff is +/- 5% on the CB, or with the alternative equation, 10%. This is often exceeded with very dilute waters and brines.
m = 2.5
x 10-5 x TDSm =
1.6 x 10-5 x specific conductance (mmho/cm)This is not as well developed as for electrolytes, and is based on empirical equations such as
log
g = ks m
ks = 0.132
m = 0.05
log g = 0.132 x 0.05 = 0.0066
g = 1.02
so as the ionic strength increases, the activity coefficient increases.
REVIEW OF IMPORTANT EQUATIONS:
1. Mass Action aA + bB ---> cC + dD
2. Free Energy G = H - ToS
D
G = (SuiGi)products - (SuiGi)reactantsD
G = DH - T°DS (at constant T,P)D
G = DG° + RT lnQ
0 =
DG° + RT ln Kor
D
G°= -RT ln KD
G = -RT ln K + RTlnQ= +RT ln Q/K
3. Temperature effects 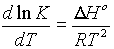
or
4. Gas Laws Raoult Pi = XiP°i
Hanks Pi = KiXi
5. Activity mi = m°i + RT lnai
and ai = giXi
6. Activity Coef. ai = gi Xi or {i} = g[i]
7.